Entretien avec le docteur May-Levin, médecin conseil bénévole à la Ligue contre le Cancer (08/03/2010) :
Présentation de la Ligue contre le Cancer
Cette association regroupe 103 comités départementaux partout en France et constitue un seul organisme indépendant à lutter contre le cancer. Elle s’axe sur trois objectifs : la sensibilisation à la prévention et au dépistage par l’information, l’accompagnement des malades et de leurs proches dans leur combat, et soutien actif à la recherche. La ligue est d’ailleurs le premier financeur associatif de la recherche contre le cancer.
Historique et cadre juridique :
Depuis qu’il est Homme, l’Homme cherche. Mais dans quelles conditions ?
Louis XIV souffrait d’une fistule annale, avant de l’opérer le chirurgien a testé différentes méthodes sur des mendiants de la ville souffrant du même problème. Autant dire qu’il n’y avait aucune éthique.
Il faut attendre le drame de la Shoah, et les abominations du docteur Mengele pour que les essais cliniques soient encadrés. Ce sont les directives de Nuremberg qui fixent en 1945 tous les grands principes éthiques encore en vigueur aujourd’hui. Le code Nuremberg vient formaliser ces directives en 1947. Organisé en dix points, ce code insiste sur le caractère primordial du consentement volontaire du patient à la recherche. Il stipule également que la recherche doit avant tout apporter du bien à la société et que les risques pris ne doivent pas outrepasser les bénéfices attendus : c’est l’être humain qui doit être mis au cœur de la recherche. Le code de Nuremberg est complété par la déclaration d’Helsinki en 1964 puis par différentes directives européennes.
En France, la première loi encadrant la recherche est passé en 1988. La loi Huriet-Serusclat sécurise les conditions de la recherche clinique : le point clé de cette loi est l’information. Les patients doivent être informés des risques qu’ils encourent autant que des bénéfices attendus. Leur consentement doivent être écrits et ils ont le droit d’interrompre l’essai sans préjudices aucun sur le traitement de sa maladie. La loi détermine également les obligations du promoteur de l’essai et des médecins investigateurs : transparence et loyauté leurs sont explicitement demandées.
La loi était au départ assez mal acceptée par les médecins à l’époque car les médecins avaient peur de dire à un malade qu’il a été tiré au sort pour participer à un essai Actuellement cette obligation d'information loyale et complète est parfaitement acceptée. Et les médecins réalisent que les patients préfèrent être vraiment informés. Un médecin ne respectant pas la loi encourt jusqu’à cinq ans de prison. Aujourd’hui,
Définitions et déroulement des essais:
La recherche clinique obéit à des conditions rigoureuses, reste la seule méthode d’évaluation objective des techniques nouvelles. C’est au nom de cette objectivité que les essais sont « randomisés », c’est-à-dire que les patients sont tirés au sort afin de comparer le nouveau traitement au traitement de référence. La recherche clinique est pluridisciplinaire, elle fait intervenir des domaines aussi divers que : chimie, pharmacologie, biologie, statistiques, cinétique,…
Elle ne peut être menée qu'après une longue phase d'étude préclinique :durant laquelle les tests sont menés uniquement sur des animaux ou des cellules sur lesquels les chercheurs déterminent les propriétés de la molécule et la dose maximale à partir de laquelle les effets toxiques apparaissent.
Les véritables essais cliniques commencent alors. Ils se déroulent en quatre phases :
- Phase I: La phase de tolérance : cette phase doit conduire par augmentation successive des doses à déterminer chez l’homme la dose limite de toxicité et la dose maximale tolérée, à savoir sans effets secondaires. En cancérologie, ce sont souvent les patients chez qui plus aucun traitement ne peut être entrepris qui se prêtent à ces essais. Et dans certains cas, ils peuvent en obtenir un réel bénéfice
- Phase II: Cette phase doit déterminer l’efficacité clinique du médicament. On teste le médicament sur un grand nombre de patients atteints de cancers différents et si l’on constate un taux de rémissions supérieur à 50%, les essais peuvent continuer.
- Phase III On passe alors à une phase de comparaison du nouveau traitement avec le traitement préexistant le plus efficace tout en étant bien toléré. En effet, en cancérologie, il est impensable de faire des essais placebos. On regarde deux choses : le traitement est-il plus efficace et/ou est-il mieux toléré ?
- Enfin, on entre dans la phase de pharmacovigilance qui débute à la commercialisation du nouveau traitement et n’est a priori jamais close. On observe et signale aux autorités compétentes les effets secondaires à long terme.
Ces essais sont donc menés dans la sécurité et la transparence absolue par l’intermédiaire d’une grande rigueur scientifique. L’OMS détaille ainsi tous les critères de rémissions dans le cas du cancer.
Les essais sont très largement détaillés dans un protocole qui définit : les objectifs primaires et secondaires de ces essais, les arguments scientifiques qui conduisent à leur mise en œuvre, le nombre de patient à inclure, les bénéfices attendus, les risques encourus, les méthodes de mesure préconisées,…Il comporte un important versant juridique pour les essais venant des laboratoires américains. Ce protocole est strictement confidentiel.
C’est le promoteur de l’essai qui est chargé de rédiger et de transmettre le protocole. Le promoteur est celui qui propose l’essai, qui prend les assurances, et qui se tourne vers les investigateurs de son choix. Il se charge de l’organisation matérielle et de la coordination des essais : tous les frais occasionnés sont à sa charge. Ce sont des industriels à 90%, les 10% restant étant des personnes morales, fédérations, états surtout engagés dans les essais de techniques nouvelles.
Les essais sont menés par des investigateurs regroupés en centres de lutte qui coordonnent tous les essais. Ce sont des médecins qui ont une double obligation : scientifique et humaine.
L’expérimentation sur les animaux et les cultures de tissus dure de deux à quatre ans puis les essais durent de cinq à six ans. Un nouveau traitement met donc plus de dix ans à être commercialisé : les enjeux financiers et médicaux sont vertigineux !
Validation des essais et publication des résultats
Les essais ne peuvent démarrer que lorsque le protocole a été validé par l’AFFSAPS et par un CPP, Comité de Protection des Personnes. Ces deux entités s’engagent à la fois comme contrôleur éthique et scientifique.
Le CPPP, il a pour mission de juger du contenu et de la forme de l'essai. Le statut des CPP a évolué à ce sujet : son rôle est passé de consultatif à décisionnel. Les CPP disposent donc désormais d’un droit de veto.
A la fin des essais, l'AFSSAPS est chargée de générer l’AMM, Autorisation de Mise sur le Marché.
Les comités de protections des personnes sont en fait des comités éthiques qui se réunissent tous les mois à l‘occasion de réunions de trois ou quatre heures. Ils se composent de spécialistes et de non spécialistes représentants des usagers, souvent des patients ou des proches de patients : médecins (généraliste et hospitalier), chercheurs, associations, pharmaciens, psychologues, biostatisticiens, juristes. La demande officielle est de 16 membres, dans les faits ils peuvent se composer de vingt personnes.
Ces comités sont riches de leur diversité, ils traitent pour le cas de la France majoritairement des essais de cancérologie. Très peu d’essais sont réalisés sur des bénévoles sains puisqu’en cancérologie il n’y a pas d’essais placebo. Les patients sont soumis à des examens très fouillés sur le plan médical.
Il existe à l’intention des patients une note d’information et un consentement éclairé à donner. Les essais sont rarement reçus du premier coup. Les membres des CPP sont bénévoles et le recrutement est très délicat. Il existe des titulaires et des suppléants. Lorsque les essais sont destinés aux enfants, le comité doit inclure un pédiatre et davantage de représentants des usagers. Il en existe aujourd’hui une quarantaine en France mais certains CPP manquent de membres : les volontaires sont rares et les conditions de recrutement difficiles. Parmi les désagréments liés à la gestion des CPP, on trouve les fonctions administratives que doivent gérer les bénévoles, la nécessité d’avoir des rapporteurs, l’absence de rémunération des membres. Les CPP sont financés par la DRASS et le bénévolat est une volonté du pouvoir public pour conserver l’indépendance des comités vis-à-vis des laboratoires. Il faut que chacun puisse garder sa liberté d’appréciation : pas de possibilité d’être acheté.
Le CPP peut faire comparaître le promoteur pour lui laisser la possibilité de s’exprimer. Quand l’essai est clos, il doit y avoir publication et information des résultats aux malades par une loi récente.
Malheureusement, la publication des résultats au grand public n’est pas systématique et obligatoire. Un essai non concluant ne sera pas publié et sera sans doute reconduit des années plus tard au détriment des patients. A l’inverse si les essais sont concluants, la diffusion sera très rapide et globale.
Le Comité de Patient pour la Recherche (CPR)
Ce comité fait la fierté de la Ligue contre le Cancer qui est aujourd’hui la seule association à l’avoir mis en place.
Le CPR compte une quarantaine de malades, ou proches d’anciens malades. Il est là pour informer les patients et pour s’interroger sur la forme et les moyens d’information dont disposent les patients... Le comité analyse donc, avant même les CPP, tous les protocoles d’essais liés à la cancérologie et juge de leur lisibilité et de leur loyauté. 75 % des promoteurs acceptent des critiques de la part du comité de patients, et les modifient en conséquence. Le comité rédige ainsi une feuille de lecture, sorte de résumé des protocoles, à l’intention des patients et organise des réunions d’information deux fois par an depuis 11 ans maintenant. Il dispose par ailleurs de deux journaux, trait d’union et Coté Patients, destinés aux patients qui paraissent tous les trois mois. On y recense les résultats des essais en cours.
Le bilan est positif : aujourd’hui l’information et la communication avec les patients se sont considérablement améliorées. Les patients sont de plus en plus nombreux à vouloir participer aux essais, d’autant plus qu’ils sont parfaitement informés.
Le cas particulier des enfants
Les diverses lois tiennent compte du statut différent des enfants et de tout autre personne fragile. Leur protection doit être maximale et les essais ne doivent être menés que si le bénéfice attendu est direct.
En général, les laboratoires ne font pas de recherche sur des traitements destinés uniquement à l‘enfant. Ce sont des médicaments adultes qu’on délivre en plus petites doses. Les essais thérapeutiques sont donc très rares dans ce domaine. Cependant, les associations de parents d’enfant malade se sont battues pour encourager la recherche pédiatrique. Le problème est le même pour les maladies orphelines.
Des risques de biais ?
Dans la recherche, on engage des personnes malades qui doivent être protégées et informées et font l’objet de recommandations et de réunions. Les biais ne peuvent donc être tolérés au moins sur le plan scientifique.
Pour éviter les biais, on travaille en équipe avec les patients, les médecins, les psychologues, les statisticiens. Le statisticien évalue le nombre de malades à inclure dans les essais et estime le nombre de vie que l’on pourrait sauver ou améliorer grâce à un nouveau traitement. On protège davantage les patients plus fragiles : les enfants, les majeurs protégés, les sujets âgés.
Pourtant :
L’éthique est-elle toujours respectée par les médecins ?
Les médecins veulent inclure beaucoup de malades dans les essais car les laboratoires payent les médicaments. Les malades sont donc souvent mieux soignés et mieux suivis s‘ils participent à des essais.
Certains essais sont menés dans d’autre pays européens et l’on ne dispose pas forcément de moyens de contrôle sur ces derniers. D’autres pays comme la Chine et le Japon sont totalement indépendants : ils mènent leur propre recherche.
Une solution d’avenir ?
Actuellement se développe un nouveau type de recherche : la recherche translationnelle qui vise à rapprocher la recherche fondamentale de la recherche clinique. Par exemple, on envisage de ne plus sélectionner les patients en fonction de leur maladie mais en fonction de certaines anomalies génétiques pouvant permettre de prédire les réactions des patients aux diverses thérapeutique s, ce qui permettrait une grande avancée clinique.
 Entrer dans la controverse
Entrer dans la controverse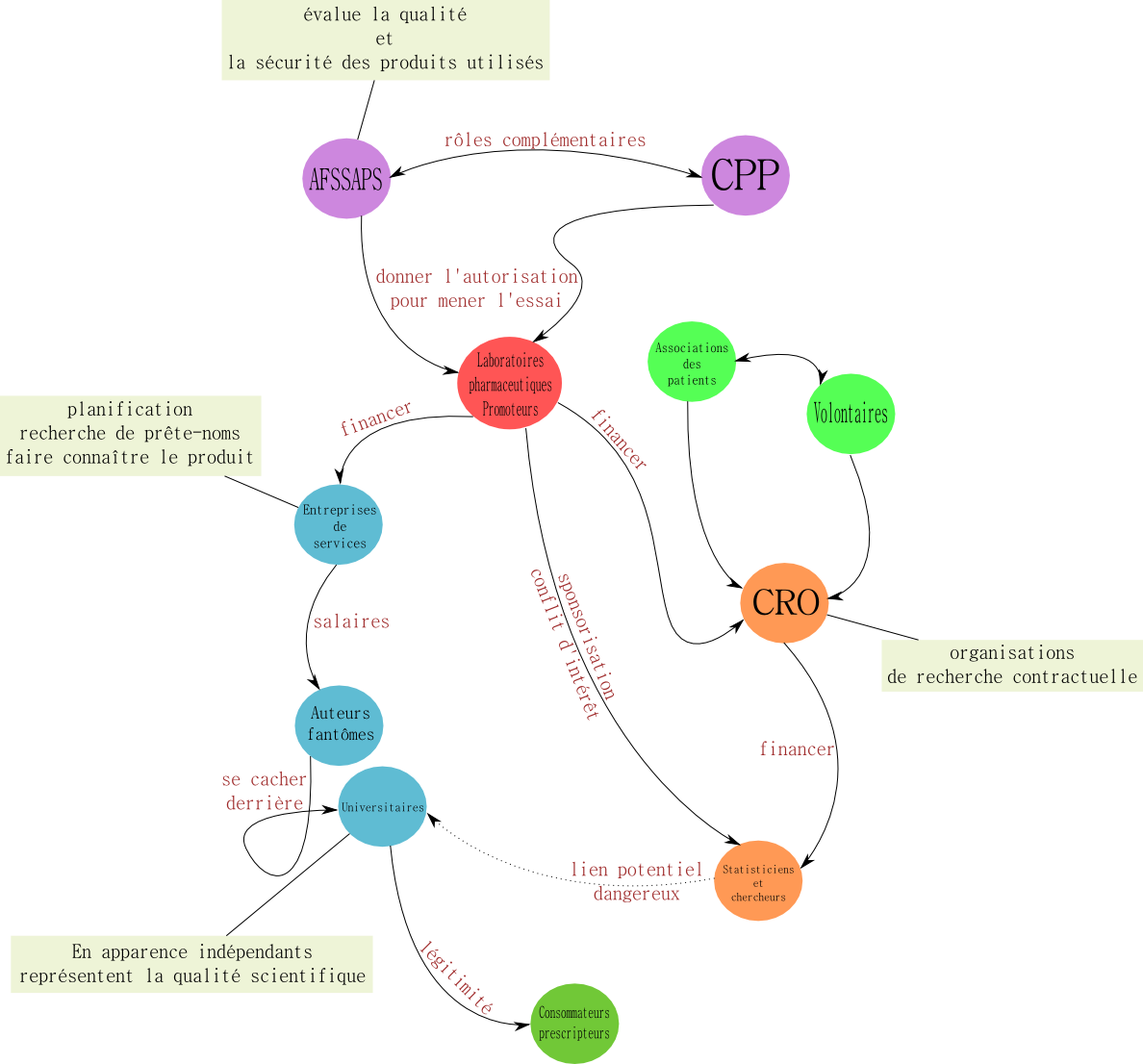
 Retourner dans la controverse
Retourner dans la controverse