• Qu’est-ce qu’un essai clinique ?
Il est difficile d’établir, à partir des différents discours scientifiques actuels, une définition unique et universelle d’un essai clinique, vu que la définition ne peut presque jamais se dissocier de la finalité de l’essai et des modèles statistique et thérapeutique adoptés pour le mettre en place. Cependant, on peut retrouver dans la diversité des approches et des avis, une sorte de principe général qui constituerait une définition toujours valable d’un essai clinique.
Un essai clinique ou essai thérapeutique est toute étude scientifique réalisée en thérapeutique médicale afin d’évaluer la sécurité et l’efficacité de l’emploi d’une méthode diagnostique, d’un protocole ou d’un médicament. Cette étude biomédicale est menée auprès d’individus volontaires sains ou malades selon le cas, après avoir réussi la phase préclinique qui évalue la sécurité du médicament en l’administrant à l’animal et en déduit la pertinence de la phase clinique, et nécessite pour ce faire l’accord des autorités de santé et des comités d’éthique qui régulent la recherche biomédicale. La définition de l’ANSM, gestionnaire et régulateur des essais en France, inclut également les études visant à établir ou vérifier des données pharmacocinétiques (modalités de l'absorption, de la distribution, du métabolisme et de l'excrétion du médicament), pharmacodynamiques (mécanisme d'action du médicament) d'un nouveau médicament ou d'une nouvelle façon d'utiliser un traitement connu.
L’objectif principal est en général d’étendre la connaissance scientifique de l’être humain et les moyens susceptibles d’améliorer sa condition. Quand il s’agit d’un nouveau médicament ou d’une nouvelle molécule, une Autorisation de Mise sur le Marché (AMM) leur est accordée quand les essais prouvent l’efficacité du médicament.
Une des caractéristiques cruciales à la rigueur d’une telle étude est son caractère prospectif qui, selon le Comité International des Rédacteurs de Revues Médicales, doit diriger la confrontation de groupes d’intervention (auxquels on a administré le nouveau traitement à tester) à des groupes de contrôle (qui n’ont pas eu le traitement en question) afin d’étudier la relation de cause en effet d’un acte médical sur l’état de santé. Cela revient à fixer au préalable la population qui sera concernée en précisant les critères d’inclusion et d’exclusion, les paramètres qui seront étudiés à partir des données recueillies auprès des sujets volontaires et les critères de sortie de l’essai. Ainsi, un modèle statistique préalablement établi est essentiel à la validité scientifique des essais cliniques. Il s’agit, en plus, d’un modèle d’étude contrôlée qui compare, par exemple, le taux de guérison du groupe ayant reçu le nouveau traitement au groupe de contrôle ayant reçu un traitement inactif (placebo) ou un traitement de référence.
• Types des essais :
- Essais randomisés : L’affectation aux différents groupes de traitement est effectuée au hasard, par tirage au sort, assurant ainsi la comparabilité de ces groupes et évitant tout biais de sélection. C’est le schéma central des essais cliniques actuels et l’on retrouve fréquemment l’appellation essai clinique contrôlé randomisé (ECR).
- Essais en « simple aveugle » : Dans ce type d’essais, le patient ignore le traitement qu’il reçoit, alors que le médecin le sait.
- Essais en « double aveugle » : Dans ce cas, le patient et l’investigateur ignorent le traitement reçu par le patient afin d’éviter le risque de modification du critère de jugement par la subjectivité du patient ou du médecin.
- Essais « ouvert » : Contrairement à l’essai en double aveugle, le patient et le prescripteur connaissent la nature et la dose du médicament prescrit.
- Essais sans bénéfice individuel direct : Une recherche est SBID quand les participants ne peuvent espérer un bénéfice immédiat (exemple : la pratique d’une nouvelle technique d’imagerie chez un volontaire sain).
- Essais avec bénéfice individuel direct : Une recherche est à BID quand on peut en attendre un bénéfice immédiat pour les personnes concernées (exemple : efficacité d’un nouveau médicament).
Les deux derniers types rappellent l’objectif des essais qui se veulent surtout comme une mise à l’épreuve des connaissances scientifiques pour les étendre dans l’absolu, indépendamment du sort des participants malades dans des maladies dont le traitement n’est pas encore mis en place ou n’est pas encore éprouvé !
• Organisation des essais :
Les essais cliniques s’échelonnent principalement sur trois phases jusqu’à l’obtention de l’AMM. Ils sont toujours précédés d’une étude préclinique pour tester la sécurité d’emploi chez l’animal et d’en déduire la pertinence de passer à l’épreuve clinique. L’épreuve clinique se prolonge par une quatrième phase qui consiste à suivre sur le long terme les effets de la nouvelle substance sur une vaste population. Les essais cliniques s’inspirent de l’Evidence-Based-Medicine (EBM) ou Médecine Fondée sur les Faits.
Il s’agit de la première phase après les tests sur les animaux et a pour but de tester la tolérance de l’organisme humain vis-à-vis de la nouvelle substance. Cette dernière est administrée par doses croissantes à un groupe de volontaires sains afin d’évaluer sa pharmacocinétique et sa toxicité, ne serait-ce que grossièrement.
Pendant cette phase, on teste la tolérance sur un petit groupe de malades hospitalisés et on cherche à déterminer la dose optimale qui permet d’obtenir le meilleur effet thérapeutique et le minimum d’effets secondaires. Les essais à cette phase sont aussi appelés les essais thérapeutiques pilotes car ils permettent de mettre en évidence l’activité chez les patients souffrants de la maladie qui doit être traitée par la substance testée.
A ce niveau-là, on compare le traitement en question avec un schéma de traitement de référence ou un placebo. On distingue alors Placebo-controlled-trials et Active-Controlled-Trials ; le placebo étant une substance à valeur thérapeutique neutre qui n’a à priori aucun effet sur la maladie. Cette comparaison cherche à vérifier la tolérance et l’efficacité en s’appliquant à des groupes plus larges de quelques centaines, voire de milliers de personnes. Cette phase permet d’établir les précautions d’emploi et de surveiller les risques d’interaction avec d’autres produits. Si les résultats sont satisfaisants avérant l’effet pharmacologique, le médicament obtient l’autorisation de mise sur le marché.
Cependant, il existe un vrai débat éthique sur le meilleur modèle des essais et s’il faut opter pour le placebo ou non. Il n’y a pas encore d’unanimité entre les scientifiques sur le modèle le plus favorable éthiquement et même scientifiquement. Les adeptes de l’usage du placebo le défendent même quand une histoire de traitement existe, soit parce que les patients peuvent présenter un profil particulier d’effets nocifs, soit parce que cette histoire apporte parfois des traitements insupportables.
Cependant, l’usage de placebo est largement reconnu comme suspect dans des situations de vie hautement menacée ou de risque d’invalidités permanentes. En plus, certains jugent injustifiable la souffrance des patients sous placebo, surtout qu’il y a eu des cas où d’autres produits sont déjà prouvés supérieurs au placebo. Selon eux, les institutions doivent restreindre au maximum la référence au placebo dans les essais.
Les essais de cette phase sont effectués après l’obtention de l’AMM. Ils peuvent concerner la pharmacovigilance ou le développement de nouvelles stratégies thérapeutiques en association avec d’autres médicaments. L’Agence de surveillance des produits de santé (ANSM et AFSSAPS anciennement) dirige le suivi du nouveau médicament en enregistrant les données de pharmacovigilance et d’assurer l’adéquation entre indication et sécurité du produit. D’éventuelles rares complications peuvent également être dépistées à ce stade-là.
•
Les essais cliniques, sont-ils mis en place correctement ?
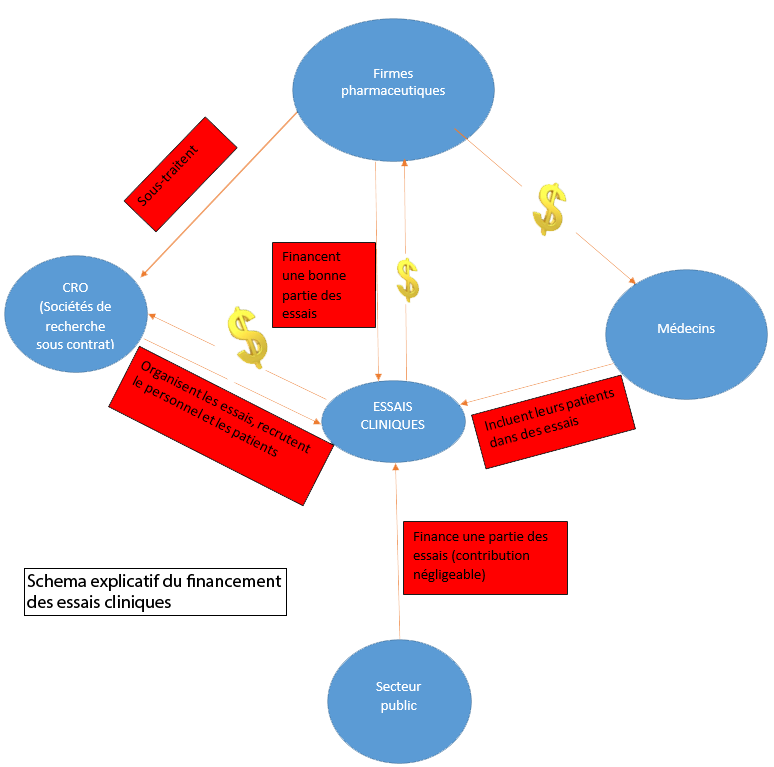
Actuellement, la grande partie des essais cliniques sont menés par les laboratoires. Ces derniers désignent les personnes responsables de la coordination des essais et notamment du recueil et de la rédaction des résultats. Il existe, entre autres, des essais particulièrement conséquents qui sont dirigés sur une large échelle régionale ; voire une échelle mondiale. Ce sont, donc, des essais multicentriques (cf. Délocalisation des essais) qui sinon n’arriveront pas à trouver assez de participants, selon le professeur Bernard Debré. L’essai est fait en demandant dans le monde entier à des médecins de traiter quelques fois une dizaine de malades, donnant quelques fois mille dans le monde entier ; ce qui donc atteste d’un manque significatif de patients par protocole, voire par mini protocole. En plus, comme les gens ne sont pas les mêmes, et que les habitants dans le nord de la France ne ressemblent pas à ceux qui habitent dans le sud de l’Afrique, l’essai risque d’emblée de ne pas être concluant, vu la grande variabilité initiale des paramètres médicaux intervenants.
« Et pourtant ils rentrent dans le même protocole. Bien souvent. Donc ce ne sont pas les mêmes. On n’arrive pas à complètement vérifier ces protocoles parce qu’il y a des malades qui ont disparu. Pourquoi ? Des fois ils sont morts. Quelque fois ils ont disparu parce qu’ils sont partis et ces protocoles la en règle général éthiquement sont très contestables, statistiquement sont ultra contestables […] parce que le suivi des patients n’est pas bien fait.» -
Bernard Debré
Le professeur souligne également le problème des essais randomisés dont l’un des bras est un placebo, en le confrontant aux essais randomisés dont l’un des bras est un traitement déjà existant. D’abord, il regrette que mis à part quelques rares cas, les double-aveugle sont obligatoires. Ensuite, il trouve très éthiquement contestable l’usage du placebo dans des pathologies qui présentent de véritables dangers sur la vie humaine et estime beaucoup moins contestable et nettement plus juste la comparaison à un traitement déjà existant mais pas forcément guérisseur.
De leurs côtés, Nicole et Gérard Delépine affirment ce qu’ils appellent la manipulation des essais cliniques par les laboratoires pharmaceutiques et se prononcent à haute voix sur la puissance économique et politique de ces acteurs (cf. Financement des essais), tout en dénonçant les différents conflits d’intérêts qui accompagnent tout la mise en place des essais (des spécialistes de signature, des professeurs d’université liés aux labos, etc.).
« Les labos n’ont rien dans leurs tuyaux. Quand je dis dans leurs tuyaux, c’est qu’il faut à peu près de 7 à 10 ans entre le moment où on pense qu’un médicament est efficace et le moment de mise sur le marché. Ils ont 10 ans d’exploitation rentable. Avant il y avait un type qui dirigeait tout ça, et qui faisait la stratégie. Maintenant c’est un financier qui dirige la stratégie. Plutôt que de faire des recherches, il préfère copier, il rajoute un hydrogène sur une molécule connue, simplement ça permet d’avoir un nouveau brevet et ça permet de repartir sur 10 ans d’exploitation […].
Le problème, c’est que c’est les labos sont propriétaires de l’essai, tout le vice vient de là, il faudrait changer le régime de propriété, je sais très bien que c’est utopique car tous les politiques qui sont achetés par les gros labos ne vont pas accepter un truc comme cela et la réalité, c’est que ça devrait être une copropriété. Deuxièmement, il devrait y avoir un enregistrement annuel des données de base, pas des résultats, mais on a aucune chance de l’avoir avec le système actuel
tant qu’on ne fera pas ça, les essais publiés seront truqués. Comment marche un essai ? Il y a des essais qui sont scientifiques, il y en a plein, heureusement, faits sur l’homme et qui sont sympathiques, parfois idiots, mais ça, on peut l’éviter. En tout cas ils sont honnêtes !» Gérard Delépine
Ce constat, à la fois de départ et d’aboutissement, fait que les docteurs Delépine ne soient pas du tout d’accord sur la manière dont sont conduits les essais cliniques à l’heure actuelle, qui est régie avant l’éthique médicale par les intérêts financiers des laboratoires de faire prospérer leurs activités de recherche et perpétuer leurs commerces pharmaceutiques. Comme les laboratoires ont la propriété intellectuelle de l’essai, ils sont propriétaires des données qu’il produit. C’est là où les laboratoires bravent les législations sur la transparence d’investigation en « truquant » les résultats des essais de façon à garantir leur mise en place de façon continue (cf. Publication des Résultats). Les laboratoires introduisent, également, par leur lobby, un biais dans les sélections des patients, dans les protocoles proposés et les volontariats appelés (cf. Consentement Eclairé), mais également des biais dans les critères de l’évaluation de l’essai et de l’exploitation de ses données. Ainsi, cette dérive, due à la globalisation et au capitalisme financier, ne fait plus prévaloir la pertinence de les molécules en question et éloigne les essais cliniques de leur mission originelle qui est de mettre au point de nouvelles techniques de traitements, contrairement au temps où les recherches cliniques étaient conduites par des instituts nationaux qui s’adressent à des marchés relativement faibles et mobilisent des agents de la santé publique sans corruption financière.
•
Les essais cliniques, mènent-ils au progrès scientifique ?
Les docteurs Delépine reconnaissent que la démarche théorique des essais cliniques est scientifiquement rigoureuse et tout à fait éthique, une fois le consentement éclairé accordé par les patients.
« Il y a un consensus dans le milieu médical pour les essais thérapeutiques, une pub permanente qui dit qu’il n’y pas de progrès sans les essais thérapeutiques…Cependant, la médecine existait bien avant les essais et la majorité des progrès ont été fait avant d’introduire les essais. On n’a pas eu besoin d’attendre les essais pour inventer des médicaments, et heureusement d’ailleurs, sinon on n’aurait pas l’aspirine, car elle provoque des ulcères chez le rat, 1870, produit qui n’aurait jamais passé la barrière des essais !» -
Gérard Delépine
Ainsi, les abus systématiques dans l’industrie de la recherche clinique, engendrent un certain scepticisme vis-à-vis de la véritable contribution des essais thérapeutiques au progrès médical sur la période des trente dernières années, typiquement. Selon Gérard Delépine, la médecine fait beaucoup moins de progrès durant les 20 dernières années qu’auparavant, en cancérologie en l’occurrence. La mise en place des essais, systématisée par les labos et catalysée par les nouvelles molécules et l’émergence des traitements génétiques, porte atteinte aux données acquis de la science en voulant en créer d’autres dans la continuité prétendue. Vu qu’il s’agit le plus souvent de « me-too », médicaments fort semblables à ceux déjà existants et ayant presque le même effet sinon inférieur, la systématisation passe par une désinformation et une automatisation de la médecine, contraires à la science et à l’éthique et contaminant tous les niveaux de formation et de soin (cf. Méthode Delépine).
Les docteurs Delépine, en se basant sur des macroanalyses de la production scientifique, nomme comme alternative la « Recherche historique » comme étant le moyen qui a toujours conduit aux percées médicales (Gérard Delépine). Ce moyen consiste en la prise en main des traitements éprouvés de la médecine dans différentes pathologies et de l’amélioration de ces techniques. Les progrès sont repérés en comparant les résultats des différentes équipes qui ont mis en place différents protocoles pour leurs patients, en partant toujours du principe que la santé du patient prévaut sur toute quête de progrès (cf. Réglementation des essais). Pour les Delépine, pour toutes les maladies, les progrès ont été fait de cette manière, considérant que les essais est une démarche beaucoup trop récente en médecine et n’intervient que pour vérifier rigoureusement les progrès inventés ailleurs au sein de la pratique médicale de tous les jours, et en comprendre les mécanismes.
« Le progrès a été fait parce qu’on a inventé une méthode, un médicament, un moyen de l’utiliser, et mieux, une technique chirurgicale. C’est ça qui a fait faire le progrès. L’essai vient juste pour l’enregistrer, prouver que ce n’est pas dû au hasard. Attention, c’est fondamental ! Quand les gens disent on ne fait pas de progrès sans essai, ce n’est pas vrai ! » -
Gérard Delépine
Mais en réponse à cette approche, Bernard Debré est d’un avis complètement différent, d’abord. En effet, selon lui, la démarche par protocoles est absolument indispensable à toute mise en forme ou coordination de tout progrès scientifique. La vision de Debré, bien en restant critique vis-à-vis des essais cliniques menés par les laboratoires dans leurs mises en place éthique et statistique contestables, est centrée sur l’essai clinique et sur le protocole de traitement. La certification (phases I, II et III) et l’évaluation à long terme (phase IV) forment une base de la construction des avancées médicales, à condition d’y respecter les modèles théorique correspondants. En plus, même les essais contre placebo, complètement inadmissibles pour les Delépine, peuvent avoir un intérêt, selon Debré, notamment pour des maladies psychiques et des pathologies infectieuses qui ne menacent pas la vie des patients. L’enjeu majeur est de traquer les conflits d’intérêts qui occasionnent des biais considérables et d’assurer des modèles statistiques rigoureux pour obtenir des données largement exploitables.
« Le seul moyen est d’avoir une organisation complètement indépendante, peut-être mondiale, et que les sanctions soient extrêmement sévères. Là donc, il faut traquer les conflits d’intérêts, il faut analyser les protocoles, analyser les résultats des protocoles absolument. Moi je me souviens d’un protocole, les allergologues disaient c’est formidable, ils ont fait un protocole. Ils ont commencé par éliminer tous ceux qui étaient vraiment très allergiques. Il ne restait plus que 150 personnes. Un protocole avec 150 personnes ?! » -
Bernard Debré
En savoir plus sur la réglementation des essais
En savoir plus sur le financement des essais
En savoir plus sur la publication des résultats
En savoir plus sur la délocalisation des essais