La bioéquivalence
On dit qu’il y a bioéquivalence lorsque la quantité et la vitesse auxquelles le principe actif est libéré dans l’organisme est identique entre le médicament princeps et le médicament générique. La bioéquivalence est déterminée par des tests scientifiques eux-mêmes encadrés juridiquement puisque selon l’article L. 5121-1 du Code de la santé publique :
Un médicament générique doit avoir la même composition qualitative et quantitative en principe actifs que le médicament de référence, ainsi que la même forme pharmaceutique et sa bioéquivalence avec la spécialité de référence doit être démontrée par des études biodisponibilité appropriés .
Précisions sur les procédures de test
La procédure de test se divise en trois étapes successives : un test de toxicité, suivi d’une vérification de la biodisponibilité se concluant par une autorisation de mise sur le marché. Le test de toxicité est effectué chez un volontaire sain par une « escalade de dose » poursuivie jusqu’à l’apparition d’effets secondaires ; il s’agit de déterminer la marge de sécurité nécessaire afin de se prévenir de l’apparition d’effets indésirables. Pour un médicament générique, ce type de test ne semble pas nécessaire puisqu’il a déjà été réalisé sur le médicament princeps : la non-toxicité du principe actif est déjà acceptée par principe.
Le test de biodisponibilité se fait sur un échantillon réduit de patients, choisi pour leurs caractéristiques similaires en masse, taille et état de santé. Le but est de déterminer les effets du médicament sur un échantillon réduit et uniforme afin d’obtenir les chiffres les plus objectifs possibles. La biodisponibilité rend compte de la vitesse et de l’étendue avec lesquelles le principe actif gagne la circulation générale et donc l’organe cible. On calcule cela en mesurant l’aire sous la courbe (AUC) de la concentration plasmatique en médicament ; les chercheurs étant particulièrement attentifs au moment t auquel la concentration maximale, Cmax, est atteinte.
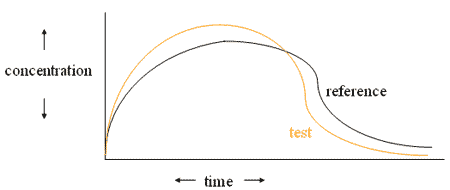
Source : onlinecourses.science.psu.edu
Une fois la biodisponibilité vérifiée arrive enfin l’étude d’autorisation de mise sur le marché (AMM). Chaque médicament, qu’il appartienne à la catégorie des princeps ou des génériques, doit être soumis cette étude pour obtenir l’autorisation à la commercialisation par les autorités sanitaires. Les génériques sont dispensés des études cliniques de toxicité puisqu’une molécule tombée dans le domaine public a normalement déjà prouvée qu’elle n’était pas nocive. Ainsi, l’AMM va être accordée après une simple étude de bioéquivalence. Ce mécanisme est expliqué dans le rapport de l’Académie Nationale de Médecine, Place des génériques dans la prescription :
L’autorisation de mise sur le marché est accordée après une simple étude de bioéquivalence, sur un groupe restreint, de 12 à 36 volontaires sains, en essai croisé (« cross-over »), comparant la biodisponibilité du principe actif du produit princeps à celle du générique après une prise unique. Les paramètres comparés sont la concentration maximum dans le plasma (Cmax) et l’aire sous la courbe (AUC). La bioéquivalence est démontrée quand l’intervalle de confiance à 90% (IC 90%) du ratio princeps/générique des valeurs moyennes de ces paramètres est compris dans un intervalle (80%-125%). Cela implique que les concentrations plasmatiques d’un générique bioéquivalant ne devront pas être différentes de plus de 5 à 7% des concentrations obtenues avec le princeps.
Le professeur Jean-Paul TILLEMENT ajoute que pour les médicaments qui ont un index thérapeutique étroit (rapport entre la dose active et la dose toxique, c’est-à-dire que la dose active se rapproche suffisamment d’une dose toxique), l’intervalle de confiance à 90% est réduit à [90%-112%].
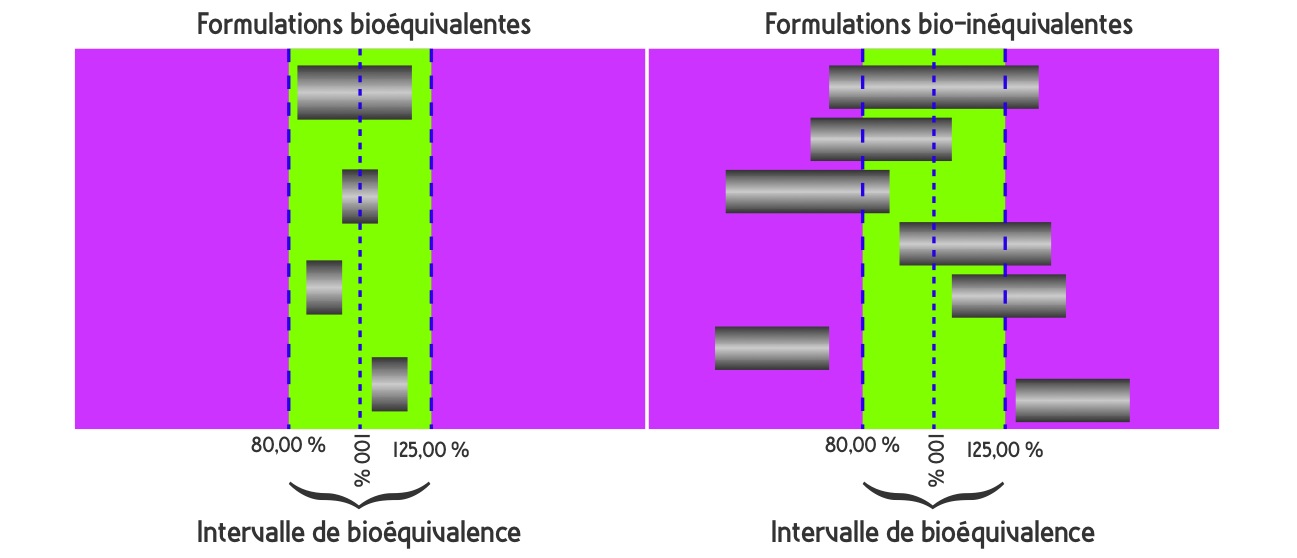
Source : Rapport de l’ANSM, Les Médicaments génériques : des médicaments à part entière. Décembre 2012
La bioéquivalence est donc une condition de commercialisation du générique. Cette procédure est très codifiée, et abondamment expliquée dans les rapports des autorités comme le témoigne le rapport de l’Académie Nationale de Pharmacie du 24 octobre 2012 « Médicaments génériques ».
Une controverse qui n’existe plus au niveau scientifique ?
Non, il n’y aucune, crainte à avoir par rapport à l’efficacité du générique s’il est bien fait.
Selon les autorités publiques, la question de l’efficacité du médicament générique a obtenu une réponse définitive au niveau scientifique. En effet, les tests de bioéquivalence sont une condition sine qua non de mise sur le marché du générique. Donc tout médicament générique contient obligatoirement la même dose de principe actif et doit donc en théorie présenter la même efficacité ; le principe actif étant en effet à l’origine de l’efficacité thérapeutique du médicament.
Cependant, les procédures de test sont remises en cause par les tenants de l’opposition aux génériques critiquant le nombre réduit de personnes testées, leur bonne santé, la prise unique d’un médicament. En résumé, les paramètres des tests ne correspondraient pas aux conditions véritables d’une prise de médicament telles qu’elle est expérimentée par un patient. Les volontaires sont choisis en nombre réduit, et pour leur ressemblance afin d’éviter toute variabilité résiduelle des tests liée aux états de santé potentiellement variable des volontaires. L’ANSM procède dans un dernier temps à des inspections régulières de bioéquivalence pour les médicaments destinés au marché français. Ces inspections se déroulent pendant la procédure d’AMM afin de valider ou d’invalider les résultats produits par les laboratoires.

La preuve par l’acceptation générale en Europe ?
Le cas français semble faire figure d’exception par rapport aux doutes qui subsistent sur l’efficacité du générique. Un des arguments des progénériques est de mettre en évidence son acceptation aux Etats-Unis et dans les pays d’Europe du Nord, la consommation y étant beaucoup plus forte. Est-ce une preuve suffisante pour montrer que les réticences des français ne sont pas fondées ?
En réalité il est difficile de faire une comparaison directe : la tradition française de la prescription sur-mesure dans les officines est différente des Pays du Nord de l’Europe qui ont toujours eu un secteur pharmaceutique plus industriel et chimique. Mais la différence majeure est celle du système de santé puisque le système des prix dans ces pays est concurrentiel, les génériques sont donc bien moins chers. Couplé à des systèmes de santé plus privatisés qu’en France, cela suffit à expliquer cette plus forte réussite des génériques.
eeeee